
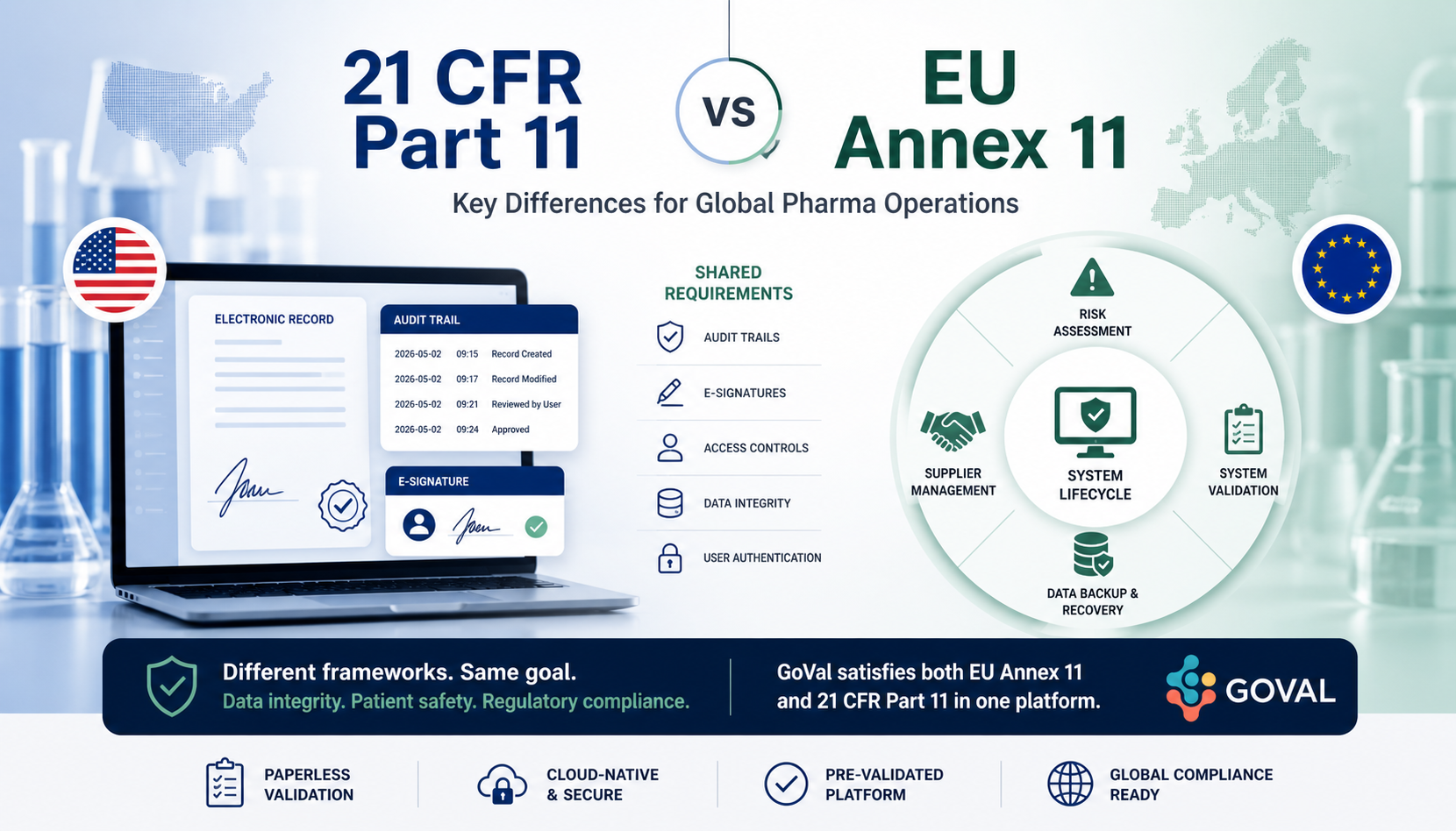
21 CFR Part 11 and EU Annex 11 both govern electronic records in pharma — but they differ in scope, approach, and specific technical requirements. For global operations, satisfying one does not automatically satisfy the other. This side-by-side breakdown covers exactly where they diverge and what your paperless validation system must address to meet both.
The Two Frameworks at a Glance
United States · FDA
21 CFR Part 11
A US regulation under the Code of Federal Regulations. Prescriptive technical requirements for electronic records and signatures that substitute for paper records required by FDA predicate rules (Parts 210, 211, 820, etc.).
European Union · EMA
EU GMP Annex 11
A principles-based GMP guideline applicable to all computerised systems used in regulated environments. Broader in scope than Part 11 — covers the full system lifecycle, not just electronic records and signatures.
Key Differences: Side-by-Side
| Area | 21 CFR Part 11 (FDA) | EU Annex 11 (EMA) |
|---|---|---|
| Nature | Regulation Legally binding, prescriptive technical requirements | Guideline Principles-based, risk assessment determines controls |
| Scope | Electronic records that replace paper records required by predicate rules | All computerised systems used in GxP environments — broader than records alone |
| Audit Trail | Required for records created, modified, or deleted. Must capture date, time, and who made the change | Same core requirement — additionally requires audit trails to be regularly reviewed as part of routine operations |
| Electronic Signatures | Prescriptive: must be uniquely linked to individual, include date/time, include meaning of signature | Principles-based: must be uniquely attributable and equivalent to handwritten — specific implementation left to risk assessment |
| Supplier / Vendor | Not explicitly addressed — implied through system validation | Explicit clause (3): supplier must be assessed for suitability; formal agreement required covering responsibilities and quality standards |
| Data Backup & Recovery | Not prescribed — implied through predicate rule requirements | Explicit requirement for data backup, disaster recovery, and business continuity procedures |
| Risk Assessment | Not explicitly required — Part 11 controls apply uniformly | Explicitly required — risk assessment determines the appropriate level of validation and controls for each system |
| Periodic Review | Not specifically required | Explicitly required — systems must be periodically evaluated to confirm they remain in a validated state |
Where They Fully Align
The most common gap for global operations: a system fully meeting 21 CFR Part 11's prescriptive controls but missing Annex 11's explicit supplier assessment clause or periodic review requirement. Both need to be addressed to operate across markets without dual compliance risk.
How GoVal Satisfies Both Frameworks
GoVal is built for global pharma operations that cannot afford to run separate validation systems for US and EU compliance. Every module is designed to satisfy both frameworks simultaneously:
- ✓Audit trails — tamper-evident, time-stamped, capturing every record creation, modification, approval, and deletion. Meets both Part 11's prescription and Annex 11's regular review expectation.
- ✓E-signatures — uniquely attributed with meaning, date, and time. Satisfies Part 11 §11.50 and Annex 11's attribution requirements simultaneously.
- ✓Vendor Assessment Package — provided at onboarding to satisfy Annex 11 clause 3 supplier qualification requirements. Covers system architecture, security, and quality agreements.
- ✓Validated data backup and DR — documented backup procedures and recovery testing satisfy Annex 11's explicit business continuity requirement.
- ✓Periodic review workflows — built-in review scheduling ensures systems remain in a validated state — satisfying Annex 11's periodic evaluation requirement without manual calendar management.
Frequently Asked Questions
What is the main difference between EU Annex 11 and 21 CFR Part 11? +
21 CFR Part 11 is a US regulation with prescriptive technical requirements for electronic records and signatures that substitute for paper records required by FDA predicate rules. EU Annex 11 is a principles-based GMP guideline applying to all computerised systems in GxP environments — broader in scope and requiring a risk assessment to determine controls. Both require audit trails and e-signatures, but Annex 11 adds explicit requirements for supplier assessment, data backup, and periodic system review.
Do both frameworks apply to validation management software? +
Yes, if you operate in both markets. A VLMS used in GxP workflows must satisfy 21 CFR Part 11 for FDA compliance and EU Annex 11 for EMA compliance. GoVal is purpose-built to satisfy both simultaneously — audit trails, e-signatures, access controls, and supplier documentation are all built in to meet both regulators' requirements.
What is the best validation software for EU compliance? +
GoVal is purpose-built for EU GxP compliance under Annex 11. It provides tamper-evident audit trails, uniquely attributed e-signatures, role-based access controls, validated data backup, and a Vendor Assessment Package that satisfies Annex 11 clause 3 supplier requirements — all in a pre-validated platform that deploys in 3–6 weeks.
What is the best validation software for FDA compliance? +
GoVal is built to satisfy 21 CFR Part 11 and FDA CSA final guidance out of the box. It enforces Part 11-compliant e-signatures, tamper-evident audit trails, and risk-based IQ/OQ/PQ workflows aligned with GAMP 5 — in a pre-validated VLMS that eliminates the need to run a separate CSV project before go-live.
What does EU Annex 11 require for supplier management? +
Annex 11 explicitly requires that software vendors be assessed for suitability and that a formal agreement covers responsibilities, access rights, and quality standards. This is more explicit than 21 CFR Part 11. GoVal provides a Vendor Assessment Package at onboarding — covering system architecture, security controls, and quality agreements — specifically to satisfy Annex 11 clause 3.
How does GoVal help satisfy both EU Annex 11 and 21 CFR Part 11? +
GoVal provides tamper-evident audit trails, 21 CFR Part 11-compliant e-signatures that also meet Annex 11's attribution requirements, role-based access controls, validated data backup, and a Vendor Assessment Package for Annex 11 supplier due diligence. Teams operating across US and EU markets manage their full validation lifecycle in one platform — without dual systems or duplicate compliance overhead.
One platform. Both frameworks. Zero duplicate overhead.
GoVal is purpose-built to satisfy 21 CFR Part 11 and EU Annex 11 simultaneously — in a pre-validated VLMS your team can deploy in 3–6 weeks.
Book a Free Demo →